Research Spotlight
Caitlin Palmer
Uncovering the Role of Mst. OB3b CopD in Copper Homeostasis
Caitlin D. Palmer,1 Rose C. Hadley,1 Madu Gamage,2 Gabriele Meloni,2 and Amy C. Rosenzweig1
1Departments of Molecular Biosciences and Chemistry, Northwestern University, Evanston, IL 60208
2Departments of Chemistry and Biochemistry, University of Texas at Dallas, Richardson, TX 75080
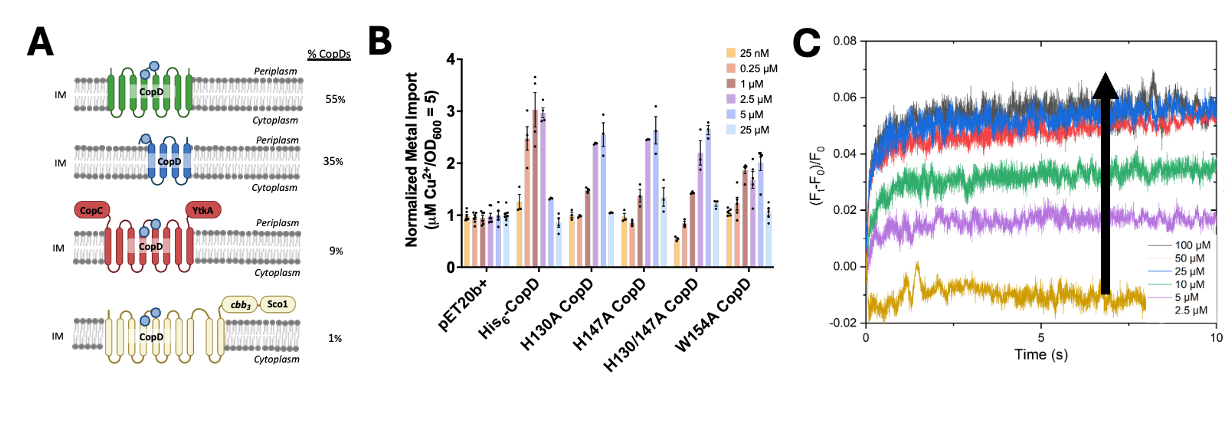
Copper is an essential cofactor for enzymes involved in respiration and redox homeostasis/defense, and thus organisms have developed elaborate copper-handling pathways to ensure proper transport and delivery to cuproenzymes while preventing cytotoxicity. Recent studies of copper transport have focused primarily on export and copper trafficking in the periplasm of prokaryotes. In contrast, the mechanisms for copper import into the cytoplasm remain uninvestigated: this process has been viewed as unnecessary since most cuproenzymes are extracytoplasmic. Increasing evidence suggests that periplasmic cuproenzymes are loaded via transport ATPases in the inner membrane following copper transport into the cytoplasm. The CopD protein family, a hypothesized inner membrane copper transporter, has been implicated along with CopC proteins in bacterial homeostasis and resistance. Here we show that overexpression of CopD from Mst. OB3b, a methanotrophic bacteria, leads to overaccumulation of copper in vivo, with a significantly higher (2-3x greater) selectivity for copper over other metals. Furthermore, two highly conserved histidine residues and one tryptophan residue located along the proposed ion conduction pathway are paramount for import, but additional soluble domains linked to the transport domain are not required, signifying a bifunctional role. In vitro studies of CopD in proteoliposomes show that the kinetics of metal transport is very fast and specific to Cu+ (K(M) = 2.13 ± 1.25 µM), and membrane potential does not build upon Cu+ translocation, suggesting a facilitated ion counter-transport mechanism. These results are consistent with the CopD family playing a role in bacterial copper import, and further characterizing this copper transport pathway will fill an important gap in knowledge regarding bacterial copper homeostasis and enzyme metalation pathways.